ARTICULO PARA COLEGAS
El Papel de los Triglicéridos en la Formación de Placas de Ateroma
Introducción
La dislipemia es la alteración en las concentraciones plasmáticas de lípidos y/o lipoproteínas. Esta alteración aumenta el riesgo de aterosclerosis porque favorece el depósito de lípidos en las paredes arteriales, con la aparición de placas de ateromas.
Aunque el colesterol elevado de lipoproteínas de baja densidad (LDL-C) está bien establecido como un importante predictor de riesgo de enfermedad coronaria y ha sido el objetivo principal de las estrategias de reducción de lípidos por el NCEP (Programa Nacional de Educación en Colesterol) de EE. UU, la evidencia sugiere que el nivel elevado de triglicéridos (TG) también es un factor de riesgo independiente.
Diferentes hallazgos sugieren que niveles altos de TG con niveles bajos de HDL-C contribuyen a un riesgo residual de sufrir una enfermedad cardio-vascular (ECV) aun cuando los niveles de colesterol LDL están en el objetivo.
Los TG, hasta donde se sabe actualmente, no son directamente aterogénicos, pero representan un biomarcador importante del riesgo de ECV debido a su asociación con partículas remanentes aterogénicas ricas en colesterol y con ApoC-III, una proteína proinflamatoria, procoagulante y causante de disfunción de las HDL.
Estructura y Metabolismo de lipoproteinas ricas en TG
Los principales lípidos del organismo son los triglicéridos (TG), el colesterol libre (CL), el colesterol esterificado (CE), y los fosfolípidos (FL). Los TG almacenados en el tejido adiposo constituyen la reserva energética más importante del organismo, el colesterol forma parte de las membranas celulares y es el precursor de hormonas esteroideas y ácidos biliares, y los fosfolípidos componen las membranas celulares y lipoproteínas haciéndolas más solubles.
Los TG están formados por una molécula de glicerol esterificada con tres ácidos grasos, son insolubles en el plasma por ello deben ser transportados junto con el colesterol y los fosfolípidos dentro de partículas esféricas denominadas lipoproteínas.
Existen cinco clases de lipoproteínas según su densidad y tamaño: QM, VLDL, IDL, LDL y HDL.
Los QM y las VLDL son las principales partículas transportadoras de TG en el plasma y son también las lipoproteínas de mayor tamaño. Los QM transportan lípidos exógenos y son sintetizados en el intestino a partir de lípidos de la dieta. Las VLDL transportan lípidos endógenos y son producidas en el hígado con ácidos grasos provenientes de: ácidos grasos unidos a albumina, ácidos grasos procedentes de los remanentes de lipoproteínas captadas por el hígado y ácidos grasos sintetizados en el hígado.
Cada lipoproteína tiene una composición lipídica, proteica y función definida. La apolipoproteína ApoB-100 es la principal proteína estructural de la VLDL y la ApoB-48 de los QM.
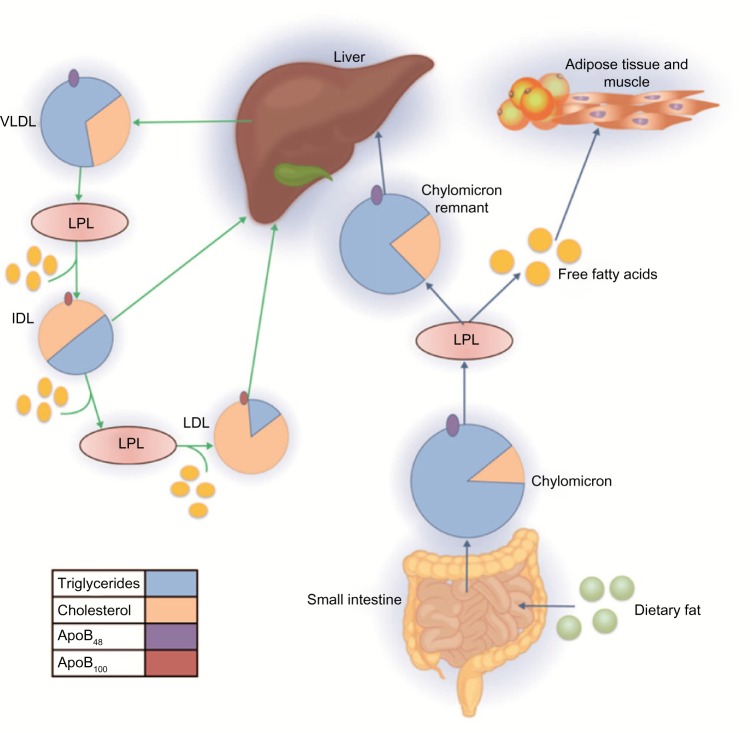
Ambos tipos de lipoproteínas van adquiriendo diferentes apolipoproteínas (ApoC-II, ApoC-III, Apo E) en circulación a partir de las HDL. A nivel de los capilares del musculo, del tejido adiposo y del corazón, principalmente, los QM y las VLDL son metabolizadas por la lipoproteinlipasa (LPL), la acción de esta enzima es hidrolizar los TG de ambas partículas liberando glicerol y ácidos grasos y originando una partícula remanente de QM y VLDL. La Apo E que está presente en la superficie de ambas lipoproteínas remanentes, funciona como un ligando para la eliminación de ambas partículas por medio de diferentes receptores: el receptor de LDL (LDL-R), la proteína relacionada con el receptor de LDL (LRP), el receptor de VLDL y el receptor de Apo E, ubicados en el hígado.
Los TG son transportados en el plasma en QM, VLDL y sus remanentes creados durante el metabolismo lipoproteico, por eso se les conoce como lipoproteínas ricas en triglicéridos (TRL). Éstas lipoproteinas son muy heterogéneas en cuanto a su tamaño, densidad, composición y riesgo cardiovascular asociado, que depende a su vez del tamaño, composición y concentración lipídica de la partícula.
Regulación del Proceso Lipolítico
Los TG no pueden atravesar libremente las membranas celulares, como consecuencia la lipolisis es un proceso esencial para la liberación de ácido grasos en los tejidos y así poder obtener energía. La LPL es la enzima clave para impulsar la hidrólisis de los TG a lo largo de la superficie luminal de los capilares y la proteína de unión a HDL glucosilfosfatidilinositol, recientemente identificada (GPIHBP1) proporciona la plataforma que permite que ocurra la lipolisis en la superficie de las células endoteliales. La LPL se sintetiza en macrófagos, adipocitos y miocitos, y debe transferirse al sitio luminal de la célula endotelial para activarse, un proceso facilitado por GPIHBP1. En condiciones de alimentación, la actividad de LPL es alta debido al efecto de la insulina, lo que resulta en una mayor absorción de ácidos grasos. La LPL está estrictamente regulada en el corazón y el músculo esquelético, ya que estos tejidos necesitan un suministro continuo de ácidos grasos para la producción de energía.
La acción in vivo de LPL está regulada por apolipoproteínas, ApoC-II, ApoA-V, ApoC-III y la proteína angiopoyetina (ANGPTL).
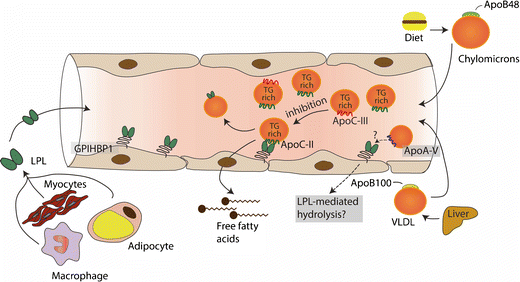
ApoC-II es un péptido de origen hepático que contiene un dominio C-terminal involucrado en la activación de LPL y un dominio N-terminal involucrado en la unión de lípidos, circula en plasma en partículas de lipoproteínas ricas en TG, así como en HDL y es la proteína limitante de la velocidad requerida para que ocurra la actividad normal de LPL. Los pacientes con mutaciones completas de pérdida de función en ApoC-II tienen hipertrigliceridemia severa similar a la deficiencia de LPL, como consecuencia, esto puede llevar a episodios de pancreatitis aguda. Por otro lado, el aumento de los niveles plasmáticos de ApoC-II se asocia con un aumento de los niveles de TG, lo que sugiere que un exceso puede tener un efecto inhibitorio sobre la función LPL.
ApoC-III es un potente inhibidor de la función LPL. Se sintetiza principalmente en el hígado y, en pequeña medida, en el intestino. ApoC-III circula en partículas de lipoproteínas ricas en TG, así como en HDL. La proteína ApoC-III se somete a glicosilación, lo que resulta en la presencia de tres isoformas que afectan su función. Estudios genéticos han proporcionado información sobre la función de ApoC- III y su relación con el riesgo cardiovascular, se descubrió que una mutación nula estaba asociada con niveles bajos de TG y aterosclerosis subclínica atenuada en la población Amish.
ApoC- III promueve la hipertrigliceridemia por diferente mecanismo: inhibición de la actividad de la LPL, interferencia con la unión de TRL a sus receptores hepáticos, por medio de la Apo B o Apo E, causando un retardo en el catabolismo de los mismos y por último, la promoción del ensamblaje y la secreción de las VLDL en el hígado. Además, ApoC-III favorece un estado procoagulante con sus posibles consecuencias aterotrombóticas. ApoC-III tiene efectos inflamatorios a nivel del endotelio mediado por la activación de la vía NF-kB, aumenta la expresión de moléculas de adhesión en las células endoteliales (VCAM-1) y en los monocitos (integrina B1), contribuyendo a la disfunción endotelial y a un mayor reclutamiento de los monocitos. Además, ocasiona disfunción de las HDL, pues las partículas de HDL unidas a ApoC-III se vuelven menos ateroprotectoras, determinándose que la concentración de ApoC-III en las HDL es significativamente mayor en personas con enfermedad cardiovascular que en personas sin enfermedad cardiovascular, esto podría asociarse con una HDL disfuncional, que no protege a la célula endotelial, recordando que la disfunción endotelial es el primer paso en el proceso aterogénico.
Las partículas HDL son: anti-inflamatorias, antioxidantes, antiapoptóticas y antitrombóticas, además participan en la protección y la reparación del endotelio. Por lo tanto, una HDL disfuncional no tendría todos estos efectos protectivos.
ApoA-V proteína de origen hepático, sus concentraciones en plasma son muy bajas y a pesar de esto su papel es esencial para el metabolismo de los TG. Variantes raras en el locus o cerca del gen que codifica para ApoA-V se asocia con niveles plasmáticos de TG aumentados y riesgo de ECV.
ApoA-V permite el acercamiento de los QM y las VLDL a la LPL, estabiliza el complejo lipolítico endotelial permitiendo la hidrolisis de los TG.
ANGPTL 3, 4 y 8 estas proteínas contienen un péptido señal, un dominio N-terminal y un dominio tipo fibrinógeno C-terminal. ANGPTL 4 se produce en tejido adiposo, ANGPTL 3 se produce en el hígado y ANGPTL 8 tanto en hígado como en tejido adiposo. Las tres isoformas pueden afectar la actividad de la LPL.
Los triglicéridos y la formación de placas de ateroma
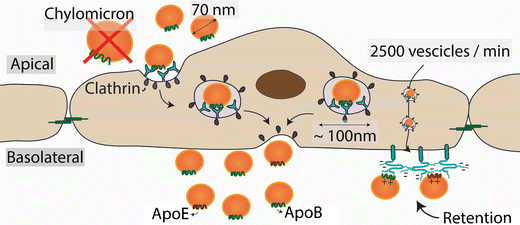
Las lipoproteínas en la circulación normalmente entran y salen de la pared arterial por transcitosis, un sistema de transporte en el que las lipoproteínas y otras macromoléculas se transportan a través de la célula endotelial en vesículas recubiertas con clatrina, estudios recientes indican que el proceso responde a los niveles de LDL en la sangre. Las vesículas de transporte tienen aproximadamente 100 nm de diámetro, por lo tanto, el sistema de transporte transcitótico está restringido a lipoproteínas de menos de 70 nm de diámetro, como son las VLDL más pequeñas, LDL, remanentes de TRL y HDL. Las lipoproteínas más grandes, como los QM y las partículas grandes de VLDL, no pueden atravesar el endotelio. Esta limitación de tamaño explica porque las personas con trastornos de acumulación de lipoproteínas grandes como los QM en paciente con deficiencia de LPL, no desarrollan aterosclerosis.
No es el influjo de lipoproteínas en la pared arterial lo que limita la velocidad y, por lo tanto, determina la concentración de lipoproteínas aterogénicas en la pared de la arteria, sino la retención subendotelial selectiva de lipoproteínas en la pared de la arteria. Dicha retención esta mediada por interacciones iónicas entre residuos con carga positiva de ApoB y ApoE en las lipoproteínas aterogénicas y grupos azúcar y sulfatos con carga negativa en las cadenas de glucosaminoglicanos de los proteoglicanos de la pared arterial, luego de ser retenidos son captadas por los macrófagos hasta convertirse en células espumosas.
Algunos autores proponen que las TRL contribuyen directa e indirectamente a la progresión de la aterosclerosis. Se proponen dos tipos de mecanismo directo e indirecto.
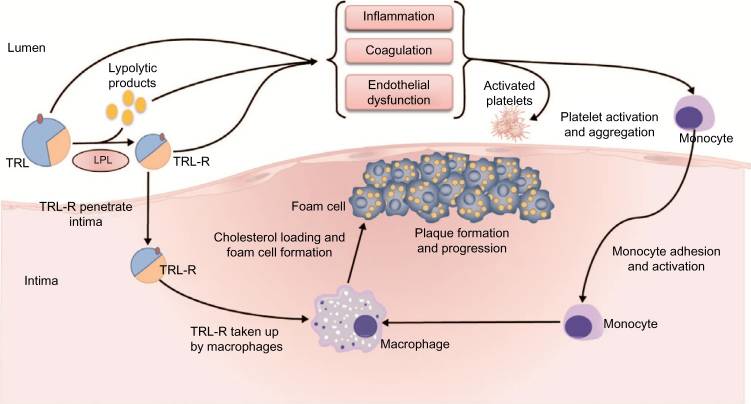
Las partículas remanente de TRL se originan cuando los QM y las VLDL son hidrolizadas de sus TG por la LPL y son posteriormente enriquecidas con ésteres de colesterol por la acción de la enzima CETP (Proteína transportadora de Esteres de Colesterol). Para evitar la cascada aterogénica inflamatoria los remanentes de TRL deben ser eliminados del plasma por medio de receptores presentes en el hígado, mencionados anteriormente. En personas con enfermedad cardiovascular las TRL permanecen aumentadas en la circulación más allá de las 4-8 horas normales. Esta falla para eliminar estas partículas se asocia a una aterogénesis acelerada, pues dichas partículas causan disfunción endotelial, de manera directa, promoviendo la expresión aumentada de moléculas de adhesión endotelial, ocasionando un mayor pasaje de leucocitos a través del endotelio para alcanzar la capa íntima arterial.
En el mecanismo indirecto la hidrólisis de las TRL por parte de la LPL ocasiona una alta concentración de productos lipolíticos, tales como ácidos grasos oxidados y no oxidados, a lo largo del endotelio vascular o dentro de la íntima arterial. Estos productos lipolíticos, junto con las TRL, activan vías proinflamatorias, procoagulantes y proapoptóticas necesarias en la patogénesis de la aterosclerosis, incluyendo un mayor reclutamiento y unión de monocitos al endotelio, una expresión aumentada de moléculas de adhesión y una vasodilatación disminuida. Los productos liberados por acción de la LPL causan daño local e inflamación en las células endoteliales, dando lugar a un endotelio disfuncional. Los sitios de disfunción endotelial constituyen las localizaciones arteriales preferenciales para la penetración y acumulación de lipoproteínas y sus remanentes y para la formación de la placa de ateroma. Las TRL pueden también suprimir los efectos ateroprotectivos y antiinflamatorios de la HDL, bloqueando el eflujo de colesterol desde los monocitos y los macrófagos.
La triada Lipídica Aterogénica
La hipertrigliceridemia también se asocia con niveles de colesterol HDL disminuidos, con partículas LDL pequeñas y densas y con partículas de HDL pequeñas.
La hipertrigliceridemia favorece el intercambio de colesterol esterificado de la HDL madura y de la LDL por TG de la VLDL por medio de la enzima CETP. El resultado es una partícula de HDL y de LDL con mayor contenido de TG y menor de colesterol esterificado. Estas partículas, con mayor contenido de TG, sufren hidrólisis por parte de la enzima lipasa hepática originando una partícula más pequeña y densa (LDL pequeña y densa al igual que HDL pequeña). Además, la HDL pequeña es removida rápidamente de la circulación, resultando en una baja concentración de colesterol HDL y un menor nivel de ApoA-I, principal apolipoproteína de la HDL, y de partículas de HDL.
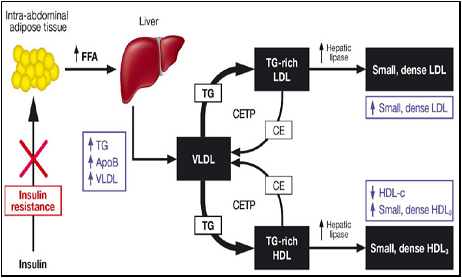
Un mayor tiempo de residencia de las TRL, que es lo mismo que decir un catabolismo retardado, brinda mayores oportunidades para el intercambio de TG por colesterol esterificado por parte de la CETP, resultando en un mayor enriquecimiento de TG de las partículas de LDL y de HDL causando una formación aumentada de partículas pequeñas y densas de LDL y pequeñas de HDL.
La combinación de colesterol HDL disminuido, predominio de LDL pequeña y densa y TG aumentados constituye la llamada ?triada lipídica aterogénica? y suele encontrarse en personas obesas, diabéticas tipo 2 o con resistencia a la insulina. Como su nombre lo indica esta condición aumenta el riesgo de ECV a partir de sus tres componentes.
El predominio de las partículas LDL pequeñas y densas está ligado estrechamente a la prevalencia de riesgo cardiovascular. La razón obedece a la mayor facilidad con que estas partículas atraviesan la pared endotelial comparadas con las partículas de LDL de mayor tamaño. Una vez dentro de la capa íntima estas LDL pequeñas se asocian a los proteoglicanos, lo que aumenta su tiempo de residencia y permite su modificación estructural, originando LDL modificadas. Además, un menor tamaño las hace más susceptibles a sufrir modificaciones. Las LDL modificadas, siendo la LDL oxidada la más conocida, es captada sin ninguna limitación por los macrófagos de la pared arterial, originando las células espumosas, características de la placa de ateroma.
Hipertrigliceridemia y nuevas opciones terapéuticas
Después de la exclusión de causas secundarias, el tratamiento de la hipertrigliceridemia leve a moderada debe seguir las recomendaciones de la guía, el paso inicial que implica el asesoramiento sobre hábitos alimenticios, tabaquismos y ejercicios etc. El objetivo en tales individuos es claramente disminuir su riesgo cardiovascular. En caso que se requiera farmacoterapia, las estatinas, los fibratos y los ácidos grasos omega 3 son todos agentes efectivos para la reducción de los niveles de TG. Actualmente se están desarrollando nuevas opciones terapéuticas que han sido basadas en evidencia genética para reducir el riesgo cardiovascular. Se descubrió una mutación nula en ApoC-III en humanos en 2008 en una comunidad Amish de los Estados Unidos que proporcionaba una Cardio protección aparente. La hipótesis del estudio respalda que la reducción farmacoterapéutica en los niveles circulantes de ApoC-III puede representar un objetivo valido para la terapia hipertrigliceridemica y, en última instancia, para la reducción del riesgo cardiovascular y potencialmente pancreatitis.
Mutaciones de perdida de función de ApoC-III se asocian con niveles bajos de TG y un riesgo reducido de cardiopatía isquémica en dos estudios de población general con más de 75000 participantes. Se hicieron hallazgos similares en el proyecto de secuenciación del exoma.
Como ya se mencionó, ApoC-III es un factor clave en la superficie de TRL y es un modulador de la actividad lipolítica de la LPL.
Sin embargo, los niveles circulantes de ApoC-III son elevados, lo que sugiere que la producción hepática de ApoC-III puede ser más viable como objetivo terapéutico en comparación con un enfoque de anticuerpos monoclonales para eliminarla, lo que permite bajos niveles plasmáticos durante largos periodos de tiempo. En este contexto el desarrollo de oligonucleótidos antisentido dirigidos al ARNm hepático de ApoC-III es muy prometedor, ya que se pueden lograr reducciones dosis-dependientes en los niveles de TG de hasta un 80%.
BIBLIOGRAFIA
- Toth, P (2016). Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vascular Health and Risk Management.
- Talayero, B and Sacks, F. (2011). The role of triglicerides in aterosclerosis. Curr Cardiol Rep.
- Luo, M and Peng D. (2016). The emerging role of opolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids in Health and Diease.
- Dallinga-thie, G., Kroon, J., and Chapman, M. (2016). Triglyceride-rich lipoproteins and remanents: target for therapy. Curr Cardiol Rep.
- Sacks, F. (2015). The crucial roles of Apoproteins E and C-III in apoB lipoproyein metabolim in normolipidemia and hypertriglyceridemia. Curr Opin Lipidol.
- Carvajal, C. (2017). Los triglicéridos y la aterogénesis. Diagnóstico clínico aplicado.
Dra. M. Victoria Gonzalez Ternavasio Bioquímica Dto. Química Clínica IBC Instituto de Bioquímica Clínica